

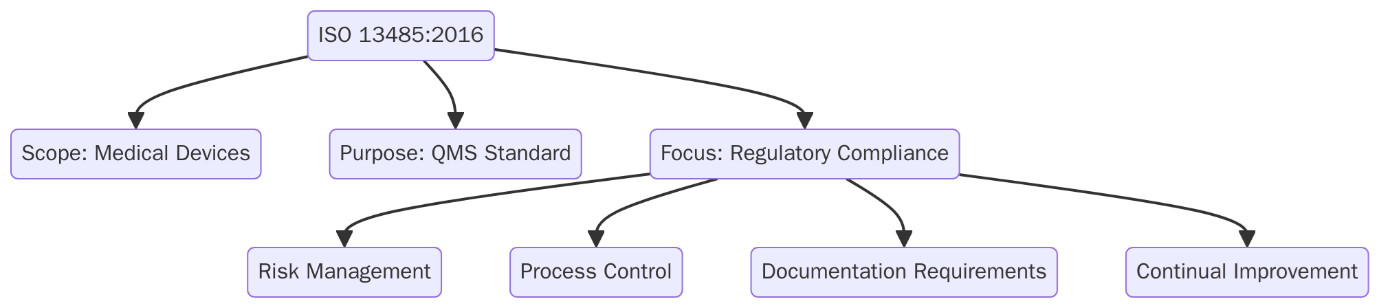
ISO 13485:2016 is the global standard for Medical Devices Quality Management Systems (MDQMS). It defines the requirements for organizations involved in the manufacturing, design, and distribution of medical devices to consistently meet customer and regulatory requirements. ISO 13485 is customized to the medical device industry, with a strong focus on risk management, traceability and compliance with global health regulations.
ISO 13485 has become more important due to increasing regulatory enforcement, the implementation of EU MDR 2017/745, IVDR and stricter post-market surveillance expectations worldwide. Moreover, authorities such as Health Canada, FDA, TGA (Australia), and Japan’s PMDA often require ISO 13485 certification for product approvals.
Whether you’re manufacturing Class I surgical tools, Class III implants or in vitro diagnostics, ISO 13485 offers a customized framework, it also supports organizations participating in Medical Device Single Audit Program.
“In today’s healthcare market, quality is not an option-it’s a regulatory expectation.” ISO 13485 helps organizations meet that expectation with confidence and consistency.
If you are looking for ISO 13485 certification, contact us at support@demo.pacificcert.com!
ISO 13485 is designed for any organization involved in the design, production, installation or servicing of medical devices. It applies across the entire medical device supply chain including those offering technical documentation or support services related to medical devices.
The standard places strong focus on risk management and keeping the quality management system (QMS) effective throughout the product lifecycle. It’s flexible and can be used by organizations of any size, whether they are directly manufacturing devices or supporting the process.
Both ISO 13485 and ISO 9001 serve different purposes. ISO 9001 is a generic standard and focuses on customer satisfaction and continuous improvement. ISO 13485 is specifically designed for the medical device industry, with a strong emphasis on compliance, risk management, and patient safety. Below are the key differences:
Feature | ISO 13485:2016 | ISO 9001:2015 |
Industry Focus | Medical devices | General quality management (all sectors) |
Risk Management | Embedded across lifecycle | Primarily at the strategic level |
Regulatory Compliance | Mandatory and detailed | Implied or optional |
Continuous Improvement | Encouraged, but less emphasized | Strong emphasis (PDCA) |
Documentation Requirements | Prescriptive (including records & SOPs) | Flexible |
Post-Market Surveillance | Required (vigilance, complaints) | Not mandatory |
ISO 13485 provides a clear framework for compliance and performance assurance. The table below summarizes clauses:
Clause | Title | Description |
1 | Scope | Defines the scope and applicability of the standard for organizations involved in the medical device lifecycle and related services. |
2 | Normative References | Lists referenced standards that are essential for the application of ISO 13485:2016. |
3 | Terms and Definitions | Provides definitions of key terms used throughout the standard to ensure consistent interpretation and implementation. |
4 | Quality Management System | Establishes general QMS requirements including documentation, quality manual, control of documents, and records management. |
5 | Management Responsibility | Outlines top management’s responsibilities for quality policy, planning, customer focus, internal communication, and management review. |
6 | Resource Management | Covers resource allocation including human resources, training, infrastructure, and work environment needed to ensure product conformity. |
7 | Product Realization | Details requirements for planning, design and development, purchasing, production, and service provision, with a strong focus on risk management. |
8 | Measurement, Analysis, and Improvement | Defines how to monitor, measure, analyze, and improve the QMS and product quality, including CAPA, internal audits, complaints, and data analysis. |
Implementing ISO 13485 involves establishing a structured Quality Management System to ensures your medical devices consistently meet safety and regulatory requirements. Below are the steps to follow:
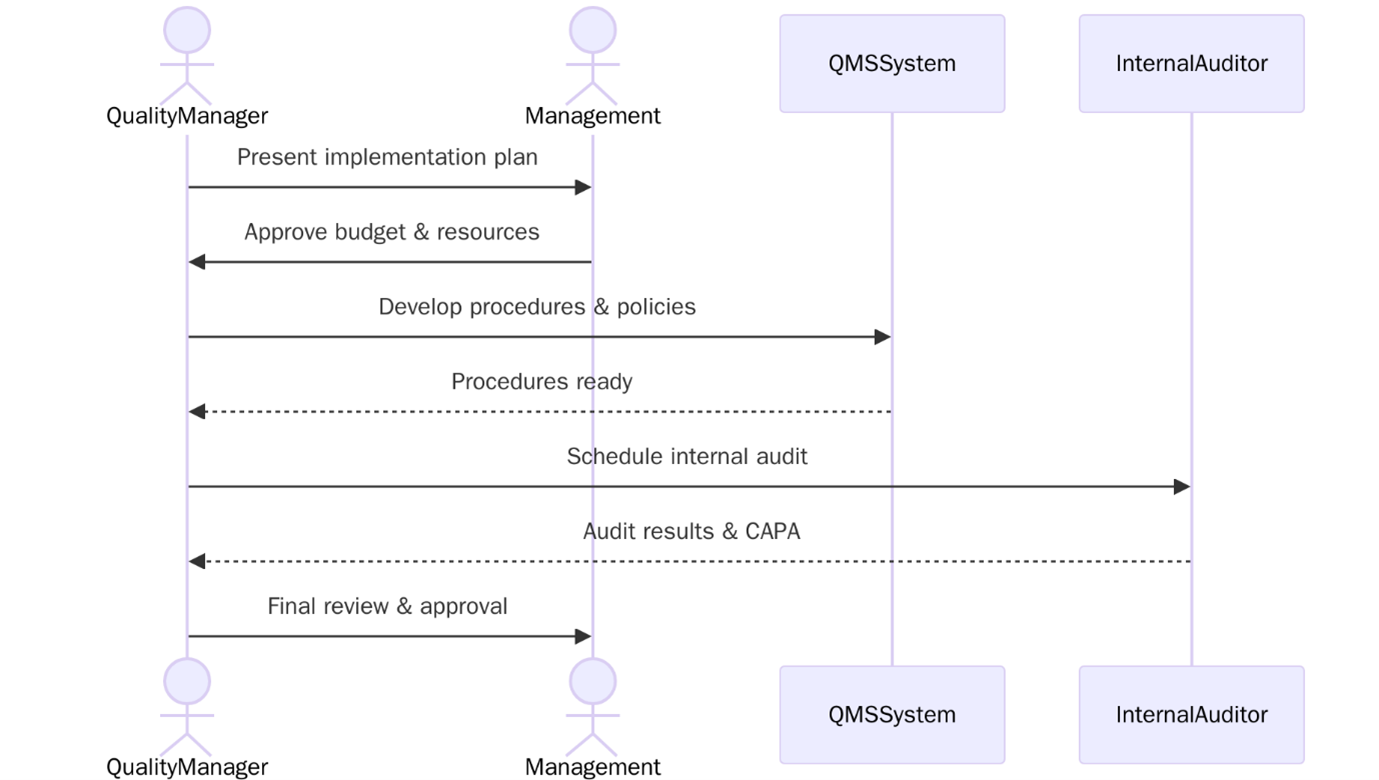
Tip: Start with a gap analysis to identify where your current processes fall short—this helps you focus resources, avoid delays, and build a compliant QMS efficiently.
The ISO 13485 audit covers key areas such as design & document control. The checklist below helps identify gaps:
Audit tip: Involve process owners during the audit, auditors want to see that staff understand and follow the MDQMS, not just that procedures exist on paper.
Understand the ISO 13485 standard and align internal goals with its core requirements, follow the steps below:
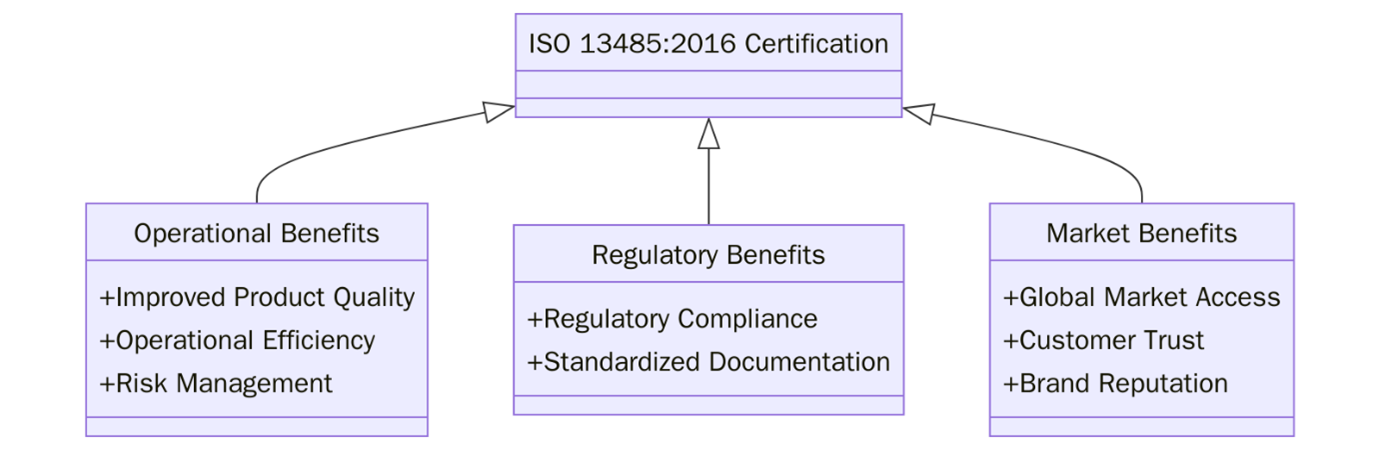
ISO 13485:2016 certification offers strategic advantages for organizations involved in the medical device sector. Below are the key benefits:
A recent global survey by Emergo by UL found that over 75% of medical device companies see ISO 13485 certification as essential for getting regulatory approval in the EU, Canada, Australia, and Japan. The U.S. FDA is also moving toward adopting ISO 13485 as the base for its updated Quality System Regulation. The medical device industry is projected to hit USD 745 billion by 2030, and certification is becoming a key factor in supplier selection.
The COVID-19 pandemic and stricter post-pandemic regulations have pushed companies to adopt stronger quality controls and risk management, making ISO 13485 a must-have for compliance and market access.
If you are looking for ISO 13485 certification audit, contact us at support@demo.pacificcert.com!
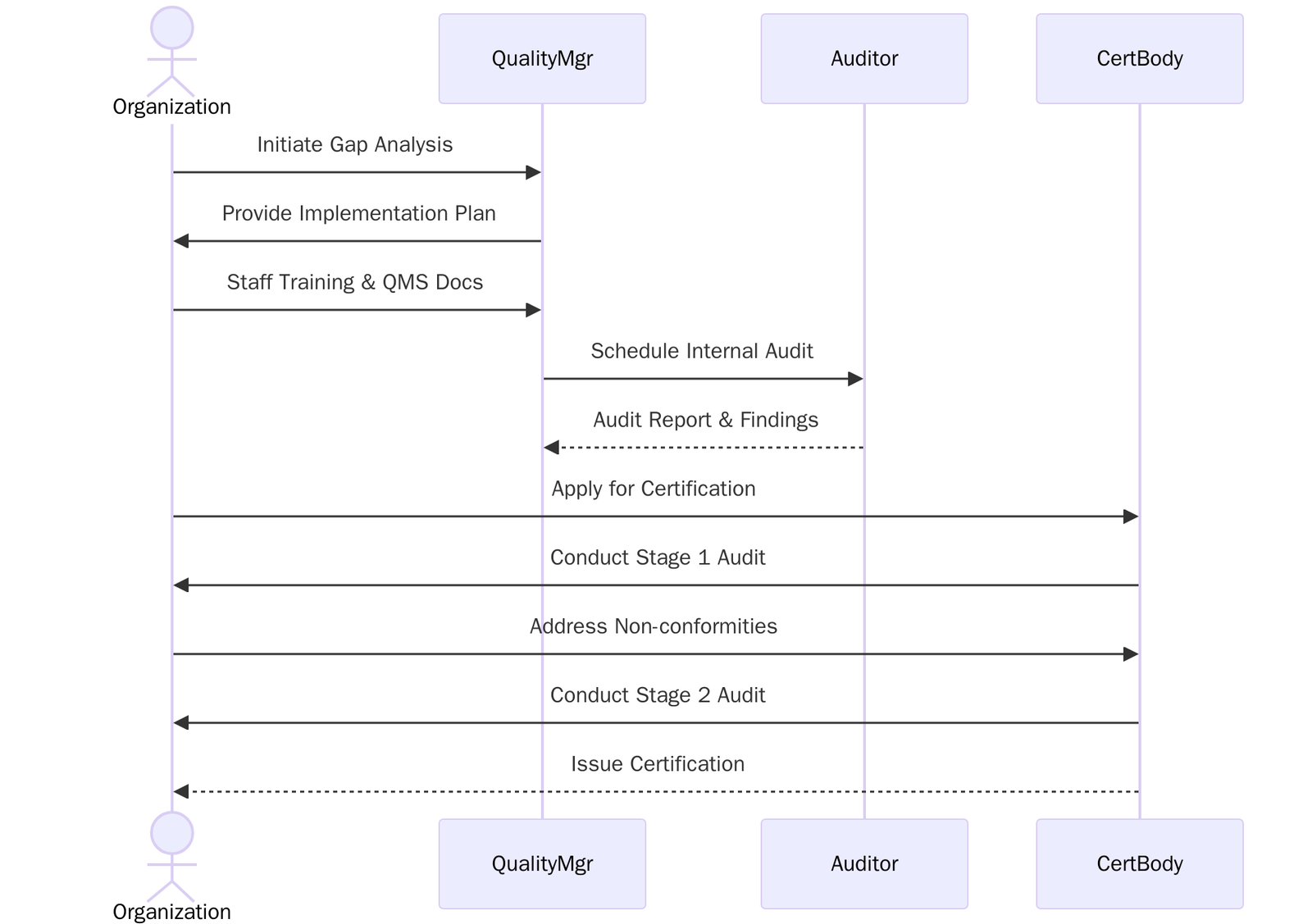
The timeline for ISO 13485 certification varies based on the size, complexity, and readiness of the organization. Below is a general timeline:
Phase | Timeline (Approx.) | Activities Included |
Preparation & Gap Analysis | Weeks 1–3 | Understand ISO 13485:2016, assess existing systems, and identify gaps |
Planning & Documentation | Weeks 4–8 | Develop QMS documents (Quality Manual, SOPs, risk files, etc.) |
QMS Implementation | Weeks 9–16 | Implement documented processes, train staff, and record evidence of compliance |
Internal Audit & Review | Weeks 17–20 | Conduct internal audits, perform corrective actions, and hold management review meetings |
Stage 1 Audit (Readiness) | Weeks 21–22 | Certification body reviews documentation and system preparedness |
Stage 2 Audit (Certification) | Weeks 25–28 | Full audit of implementation, effectiveness, and compliance with ISO 13485:2016 |
Corrective Actions (if needed) | Weeks 29–32 | Address any nonconformities identified during the audit |
Certification Issued | Weeks 33–36 | Certification body issues ISO 13485:2016 certificate upon successful audit closure |
To know your certification timeline or roadmap, contact us at support@demo.pacificcert.com.
The cost of ISO 13485:2016 certification is not fixed and varies depending on several key factors, including the size of the organization, number of employees, number of sites, and the complexity of manufacturing processes.
To receive a customized quote for ISO 13485, contact Pacific Certifications at support@demo.pacificcert.com.
The importance of ISO 13485 has grown significantly with the rise of global healthcare regulations like FDA 21 CFR Part 820 (QSR) and EU MDR. In fact, ISO 13485 is often treated as a prerequisite for market access. Without this certification, manufacturers face severe obstacles or participating in tenders.
ISO 13485 is essential for medical device manufacturers, contract manufacturers, original equipment manufacturers (OEMs), sterilization and packaging providers, distributors, and import/export agencies that deal with Class I, II, or III medical devices, including surgical instruments, implants, IVDs, and diagnostic equipment. Even software providers offering healthcare-related applications fall under its scope if their solutions are classified as medical devices.
Pacific Certifications is an accredited certification body; we offer globally recognized ISO 13485 certification services for organizations operating in the medical device industry. We conduct impartial audits that help our clients to validate compliance. We verify your system’s effectiveness and ensure compliance maintaining our accreditation integrity.
Our Services Include:
We cater to organizations across the full medical device lifecycle, ensuring your QMS aligns with requirements such as EU MDR, US FDA, MDSAP etc.
To request a quote or schedule an ISO 13485 audit, reach out to our certification team at support@demo.pacificcert.com.
Contact Pacific Certifications to begin your certification journey today!
Suggested Certifications –
Read more: Pacific Blogs
![]()
Get a rough Estimate for your Required Certification by entering your basic details.
This will close in 0 seconds
Get in touch!
This will close in 0 seconds